Lab1 Marking
You did a great job especially with the formatting. However, a wavenumber was rounded incorrectly, and you have reported wrong energy, RMS gradient, and geometric parameters. If you have any queries, please contact Prof. Hunt.
NH3 Molecule
calculation data
| Name of submitted log file |
HTHOMPSON_NH3_OPTF_POP.LOG
|
| Molecule |
NH3
|
| Method |
RB3LYP
|
| Basis set |
6-31G(d,p)
|
| Final energy |
-56.556405
|
| RMS gradient |
0.0098
|
| Point group |
C3V
|
Item Table
Item Value Threshold Converged?
Maximum Force 0.000000 0.000015 YES
RMS Force 0.000000 0.000010 YES
Maximum Displacement 0.000003 0.000060 YES
RMS Displacement 0.000001 0.000040 YES
Low frequencies --- -5.6864 -3.6131 -3.6124 0.0017 0.0048 0.0162
Low frequencies --- 1089.3674 1693.9284 1693.9284
Optimised molecule image
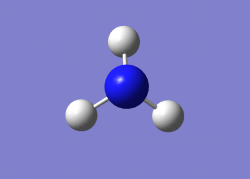
Jmol rotateable molecule
logfile: Media:HTHOMPSON_NH3_OPTF_POP.LOG
Important geometric parameters
Optimised bond distance and angle for NH3
r(N-H)=1.02Â
θ(H-N-H)=106°
Vibrations
| Mode |
1 |
2 |
3 |
4 |
5 |
6
|
| Wavenumber (cm-1) |
1090 |
1694 |
1694 |
3461 |
3590 |
3590
|
| Symmetry |
A1 |
E |
E |
A1 |
E |
E
|
| Intensity (arbitary units) |
145 |
14 |
14 |
1 |
0 |
0
|
IR Spectra

Atomic charges

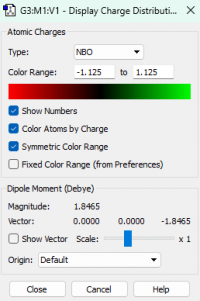
Charges of each atom
Molecular Orbital Diagram


Project Molecule
Calculation Data
| Name of submitted log file |
HThompson_N2F2_optf_pop.LOG
|
| Molecule |
N2F2
|
| Method |
RB3LYP
|
| Basis set |
6-31G(d,p)
|
| Final energy |
-309.012413
|
| RMS gradient |
0.000001
|
| Point group |
C2V
|
Item Table
Item Value Threshold Converged?
Maximum Force 0.000002 0.000015 YES
RMS Force 0.000001 0.000010 YES
Maximum Displacement 0.000003 0.000060 YES
RMS Displacement 0.000002 0.000040 YES
Low frequencies --- -0.0009 -0.0004 0.0010 3.2689 4.3149 5.0855
Low frequencies --- 347.8792 561.2465 771.6103
Optimised molecule image

Jmol rotateable molecule
logfile: Media:HTHOMPSON_N2F2_OPTF_POP.LOG
Important geometric parameters
Optimised bond distance and angle for N2F2
r(N-F)=1.3Â
r(N=N)=1.23Â
θ(F-N=N)=120°
Vibrations
| Mode |
1 |
2 |
3 |
4 |
5 |
6
|
| Wavenumber (cm-1) |
348 |
561 |
772 |
949 |
987 |
1637
|
| Symmetry |
A1 |
A2 |
B2 |
A1 |
B2 |
A1
|
| Intensity (arbitary units) |
0.6 |
0 |
75 |
75 |
81 |
21
|
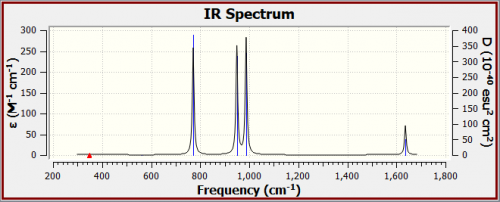
IR Spectra
Atomic charges

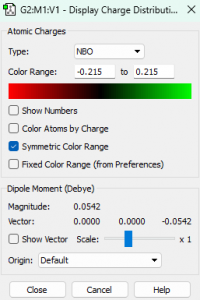
Charges of each atom
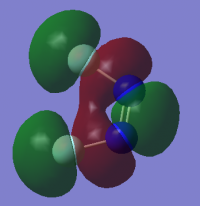
Molecular Orbital Diagram
| Molecular Orbital |
LCAO
|
 |
|
Questions to answer
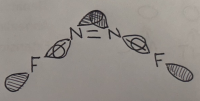
- The molecule from the log file does not have bonds between the F and N atoms, what is going on here?
- There is a preset input length that Gaussview uses for each bond, the length of the bond between the F and N atoms is too long and exceeds the preset input length. There is still a bond there however Gaussview does not show it.
- How many vibrations are expected from the 3N-6 rule?
- Vibrations = 3(4)-6 = 6 Vibrations expected
- Why are there only 4 peaks in the IR spectrum?
- The Modes 1 and Modes 2 have an intensity of 0 and 0.6. The low intensity resulted in the peaks not showing on the IR spectrum, and the 4 modes with higher intensities show up on the spectra.
- Which vibration is the asymmetric N-F stretch?
- Vibration 3 shows the asymmetric N-F stretch
- What is the nature of the highest energy vibration?
- The highest energy vibration is the N=N symmetric stretch
- Which MOs are core orbital MOs?
- MO 1-4 as they contain the different combinations of the 1s orbital phases of the 4 atoms











